Background
The In Vitro Medical Devices Regulation (IVDR) is the regulatory framework in the EU Market to harmonise safety and performance of in vitro diagnostic medical devices.
The IVDR came into force on 26-May-2017, with a 5-year transition period that ends on 26-May-2022. When the IVDR became applicable, the Directive (98/79/EC) on in vitro diagnostic medical devices will be repealed.
An Overview of Major Changesii
The IVDR presents colossal changes to the Invitro Diagnostics (IVD) industry; there will be many changes and updates to be made for the In Vitro Diagnostic Directive (IVDD) to IVDR transition. The key changes are described below.
Definition and Scope
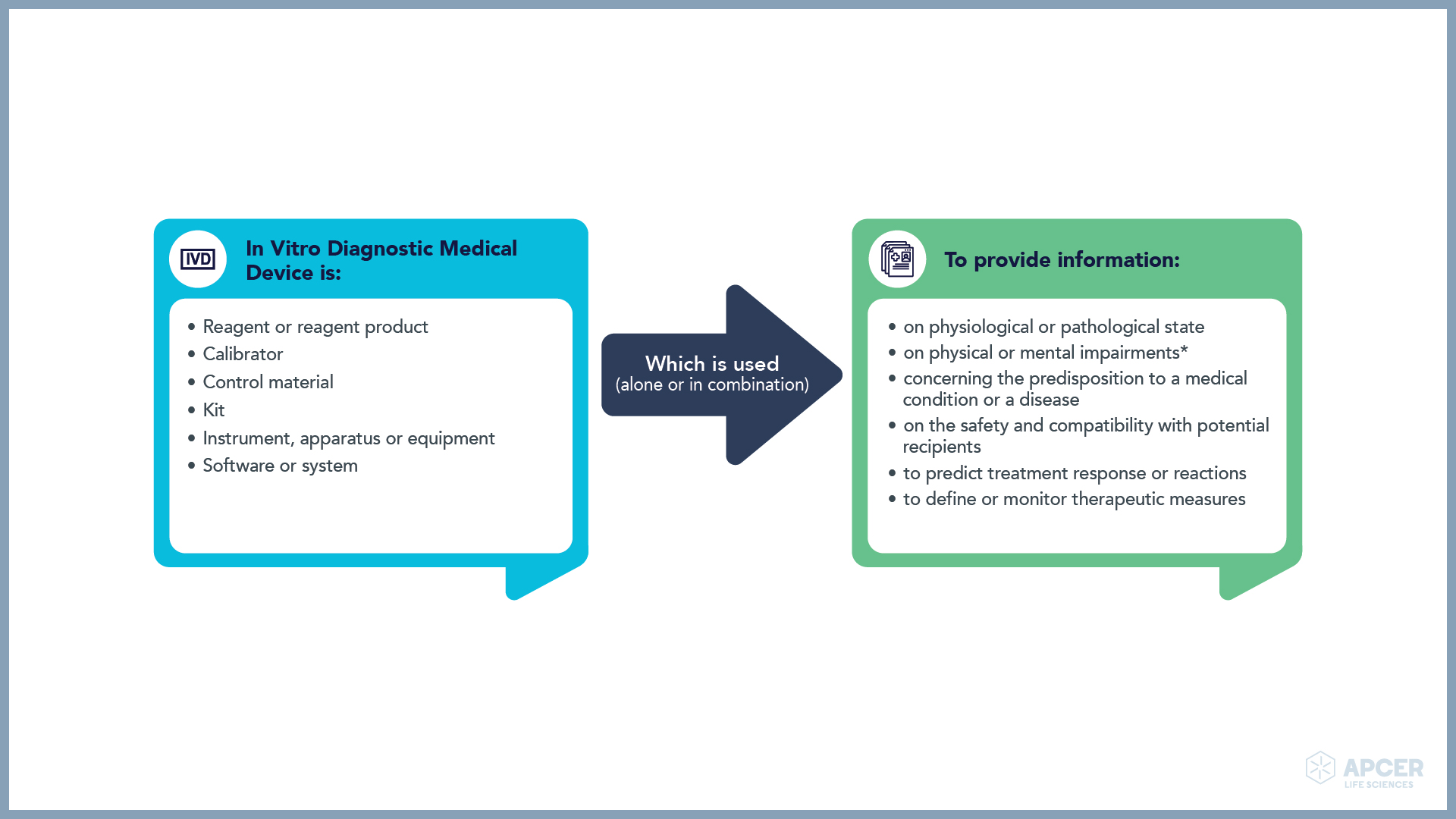
With the implementation of the IVDR, the scope has not expanded significantly; however, there are a few inclusions made in the scope, eg., high-risk in-house tests, companion diagnostics, genetic testing and other tests that provide information about a patient’s predisposition to a specific disease or susceptibility for medical treatment.
Harmonisation
This regulation ensures that all the member states have the same approach. Article 8 in the regulation states that devices that are in conformity with the relevant harmonised standards, or the relevant parts of those standards, the references of which have been published in the Official Journal of the European Union, shall be presumed to be in conformity with the requirements of this regulation covered by those standards or parts thereof.
Classification and Reclassification
The classification under the IVDR will be based on the Global Harmonization Task Force (GHTF) but this will not be identical. Therefore, interpretation of the rules will be the key.ii
Clinical evidence
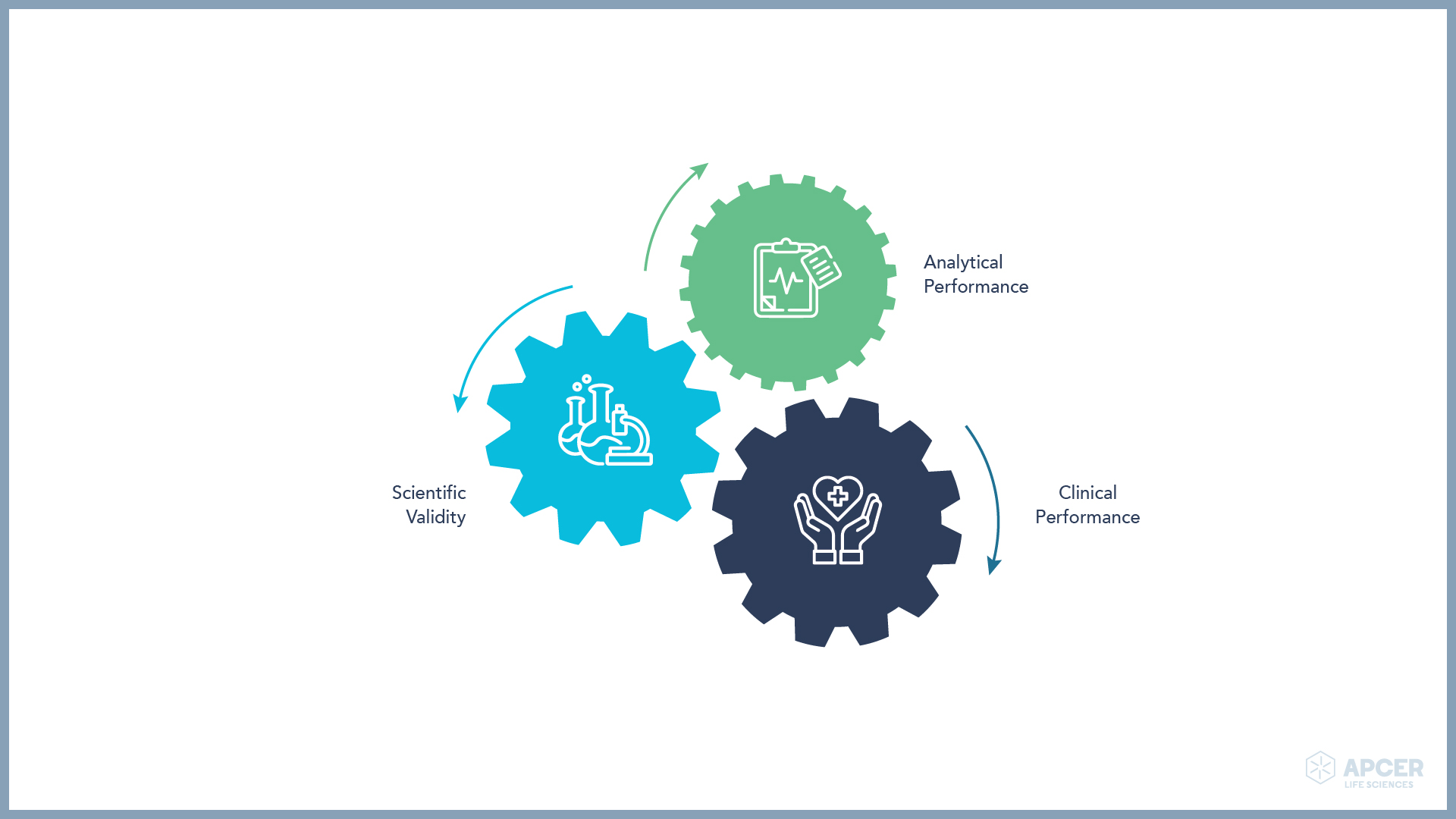
The IVDR will require clinical evidence and post-market performance follow-up (PMPF), therefore providing a life-cycle approach. Clinical evidence comprises analytical performance, scientific validity and clinical performance.
Notified bodies (NBs) and conformity assessment
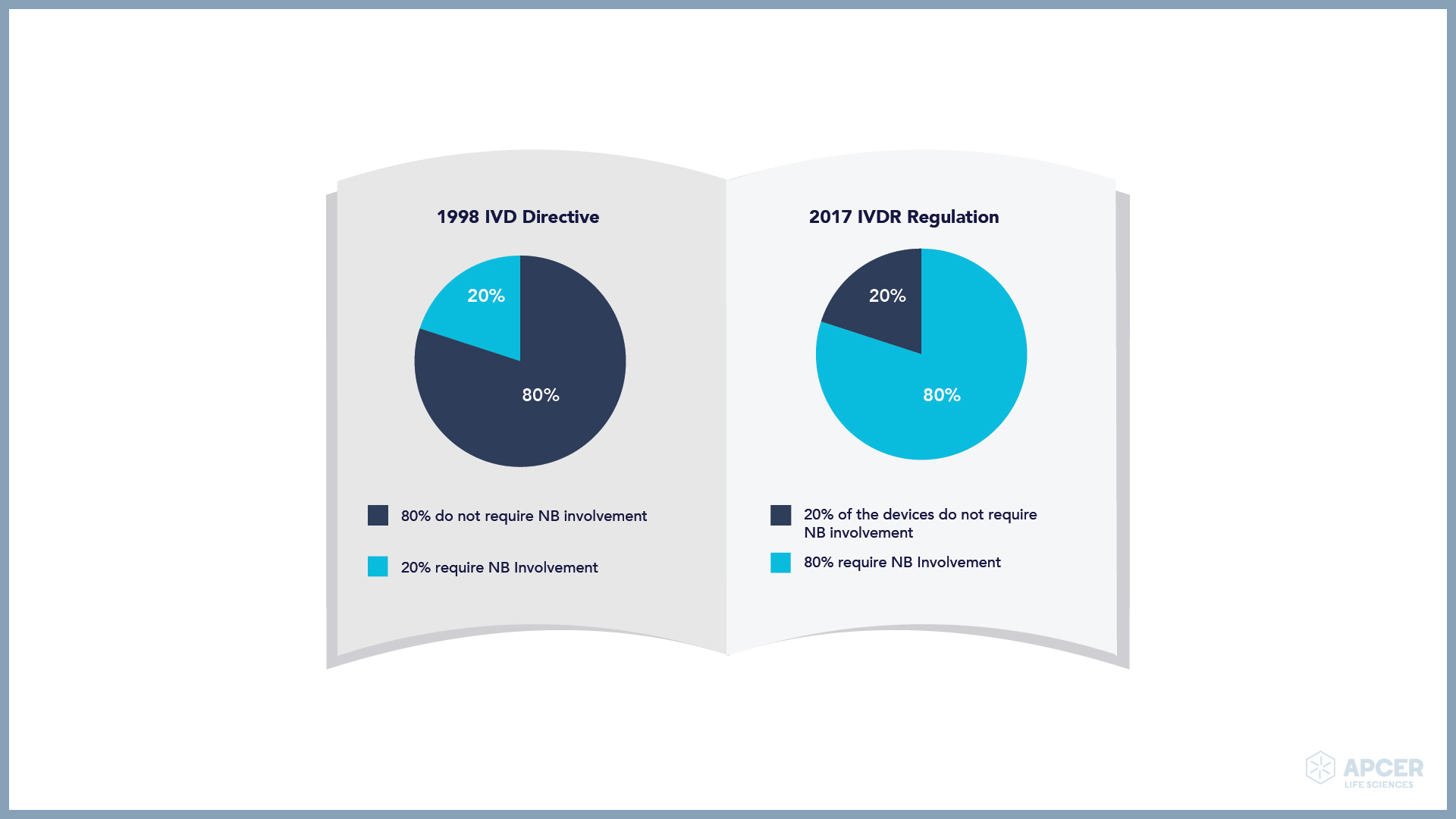
The IVD manufacturers have started assessing the product portfolio and over 80% of the devices will need an NB for the conformity assessment. Therefore, the change in the level of NB oversight is significant with the implementation of the IVDR.
Post-market surveillance and vigilance
The IVDR requires to have proactive and preventive post-market surveillance plan. All devices must have post-market performance follow-up (PMPF) plan.
Under the current regulation, manufacturers were required to conduct the post-market surveillance, whereas, under the IVDR, these requirements are more precise, binding and links to the specific new elements such as confirming scientific validity.
Technical documentation
It is important to note that the technical documentation plays a critical role in demonstrating the in vitro device performance and this does not stop at the sponsor submission, rather it evolves throughout the device life cycle. The scope and number of requirements for technical documentation has been expanded.
Quality management systems (QMS)
The IVDR has new requirements for specific plans and subsequent reports with precise content. Manufacturers of the in vitro devices shall establish a framework to continually improve a QMS to ensure compliance.
The IVDR Implementation Timeline
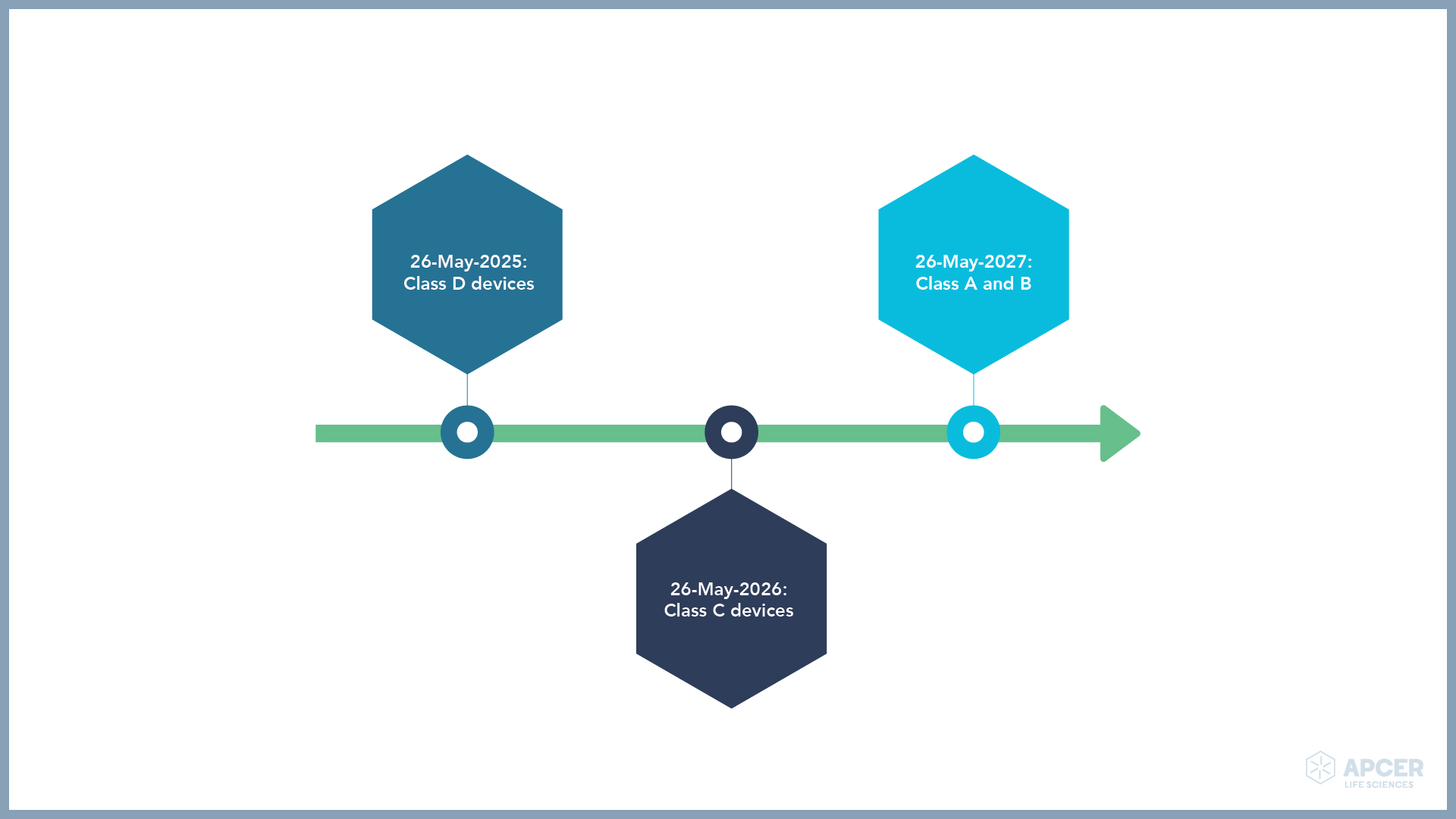
From 26-May-2022, all new certificates will have to be delivered according to the IVDR. Certificates issued under the IVDD are valid at the latest till 26-May-2024; however, post-market surveillance, market surveillance, vigilance and the registration of economic operators and device regulation will be applicable from 26 May 2022.
Devices placed on the EU market prior to 26-May-2022 as self-declared (i.e., with no NB involvement) and that require the involvement of an NB under the regulation may be placed on the market or put into service under the directive until the following dates:
How can APCER help you make your company IVDR operational?
It is of utmost importance for the IVD manufacturer to start the transition plan and avoid product availability issues. Manufacturers should recognise that the IVDR is more complex and detailed and will require more resources to maintain as well as implement. Due to the nature of changes, this will not only affect the regulatory system but all the functions in the company.
While the efforts of the majority of the staff will be directed towards meeting the regulatory bodies for product classification, CE marking, conformity routes and the scrutiny process, it may be beneficial to partner with a strategic partner to fill the gaps in implementing the IVDR regulation.
APCER’s IVDR readiness team can help your company in setting up the IVDR regulatory framework and make this operational.
Key Value Propositions
- Regulatory intelligence to keep you informed of the latest changes
- Control document gap analysis
- Procedures and templates updates to meet the post-marketing surveillance and PMPF requirements
- Assistance with technical documentation
- Training courses to support the staff of the new requirements
- Industry trends and benchmarking for the implementation of IVDR