Together for better
patient outcomes
Medical Devices
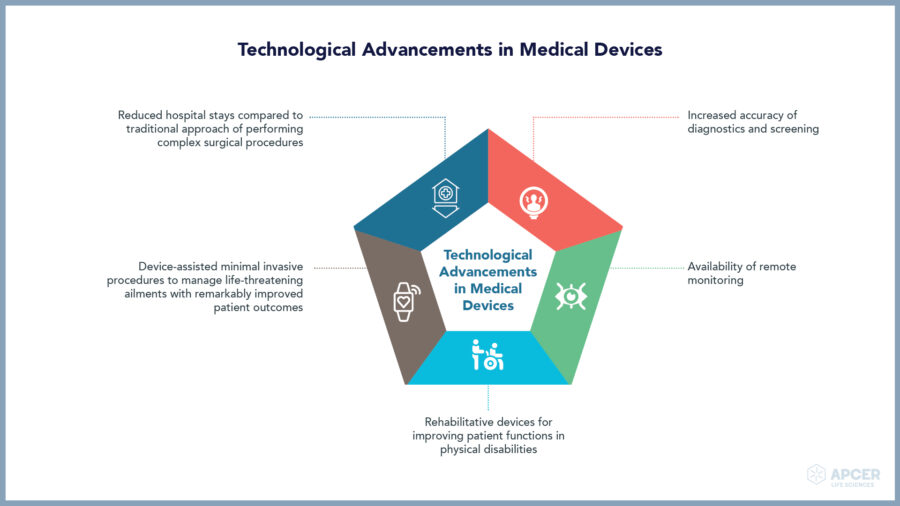
Medical devices play a key role in healthcare, vital for diagnosis, screening, monitoring, rehabilitation, treatment and care. With technological advancements, medical devices are drastically improving the efficiency and quality of health care.
Medical devices range from multi-therapies like simple tongue depressors and bedpans to complex programmable pacemakers and closed loop artificial pancreas systems. Based on the intended medical purpose, even software can also be considered as a medical device.
Medical Devices in multi-therapeutic areas

Orthopedics

Cardiovascular

Diabetes

Nephrology

Opthalmology

Oncology

Respiratory

Neurology
Key Regulatory Requirements for Medical Devices

US Medical Device Regulations
21 CFR Part 803 contains mandatory requirements for manufacturers, importers, and device user facilities to report certain device-related adverse events and product problems to the FDA

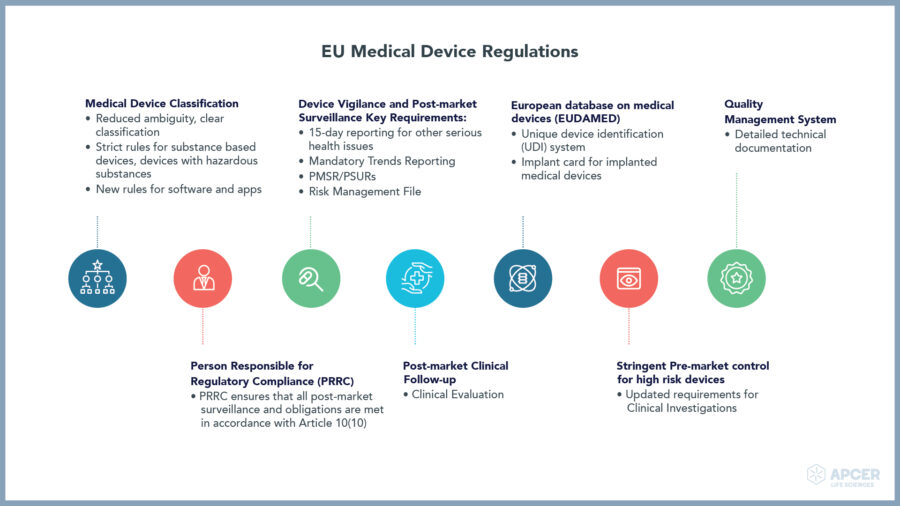
EU Medical Device Regulations
In the EU, the Medical Device Regulation (EU) 2017 /745 (EU MDR) came into force on 25 May 2017 repealing the existing Medical Device Directive 93/42/EEC (EU MDD) with transition period of four years including end of extension period in May 2021.

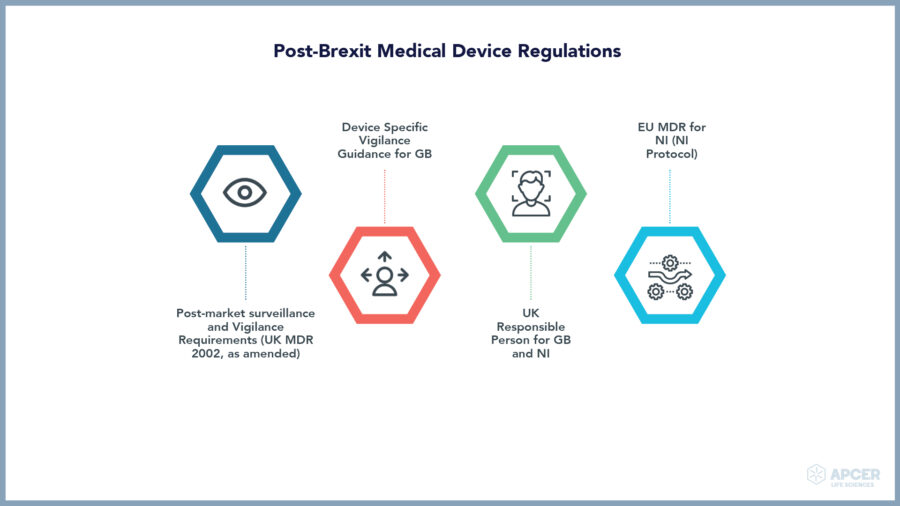
Post-Brexit Medical Device Regulations
Clinical investigations with medical devices as well as clinical performance studies with in vitro diagnostic devices are impacted, Where the single market relies on one set of rules, different rules will apply for different markets – newly defined in the EU and UK as of January 1, 2020.
Safety & Regulatory requirements for Medical Devices
The medical device companies need to have robust and effective strategies in place to ensure continued adherence to dynamic regulatory compliance and keep pace with increasing complexity within scientific elements of this area
The processes necessary to ensure compliance are complex and associated implementation activities could be challenging for small and medium organizations
- Intake of device complaints
- Intake and response to inquiries related to the medical device(s)
- Triage and flag safety reports to the safety department
End to end safety data management during clinical trials and post marketing
- Serious adverse events (SAEs)
- Unanticipated serious adverse device effects (USADEs)
- Unanticipated adverse device effects (UADEs)
- Adverse events of special interest (AESI)
- Device deficiencies
- IDE reports
- Annual safety reports
- Literature monitoring
- Trend analysis, signal detection
- Electronic Medical Device Reporting (e-MDR)/ EUDAMED
- Person Responsible for Regulatory Compliance (PRRC) as per the EU-MDR
- Authorized Representative
- Regulatory strategy design e.g., Medical device classification
- Regulatory Submissions (e.g., Technical Files/Design Dossiers, CER for CE Marking etc.)
- Expedited and periodic submissions to regulatory authorities
- Technical Documentation & Quality management systems
- Benefit-Risk Analysis and Risk Management
- Post market surveillance (PMS) plan, Clinical Evaluation and Post market clinical follow up (PMCF) plan and protocols, PMCF reports etc.
- Post market surveillance Report (PMSR)/ (Periodic Safety Update Report) PSUR
APCER Life Sciences-Your Preferred Safety Partner

Medicinal and Scientific Expertise
- 100+ physicians, 90% healthcare professionals respond to your calls, write your narratives and submit your reports to the regulatory agencies
- Expertise in handling multiple product types including vaccines, medical devices, biologics, combination products and cell & gene therapies

Client Focus
- Customized solutions through flexible operational models, strong governance and collaborative approach
- Outcome driven engagements
- End-to-end Pharmacovigilance, Medical Information, Regulatory Affairs, Quality Assurance and Medical Writing activities throughout the drug lifecycle

Global Presence
- Offices/presence in US, UK, EU and Asia
- Adverse event reporting capabilities in 100+ countries and Medical Information coverage in 50+ countries and 30+ languages, scalable to 100+ languages

Quality and Compliance
- Vast experience in supporting regulatory Inspections
- Best-in-class document control systems
- Robust QMS throughout drug life cycle